A Different Computational Biology, Bioinformatics, Biostatistics

We are interested in understanding cellular processes at molecular lever in atomic details to gain insight into functionality of molecular machines and answer what cause their malfunction, i.e. genetic and infection diseases.
To address that we develop mathematical, statistical models, using physical and chemical laws, implement them in computer algorithms and methods, programs, databases toward solving problems in biology and medicine, specifically biomedical/biotech problems involving proteins, small molecules and DNA/RNA and their interactions.
Friend, Integrated Front-End application for multiple structure visualization and multiple sequence alignment
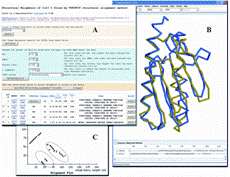
Friend is a bioinformatics application designed for simultaneous analysis and visualization of multiple structures and sequences of proteins and/or DNA/RNA. The application provides basic functionalities such as: structure visualization with different rendering and coloring, sequence alignment, and simple phylogeny analysis, along with a number of extended features to perform more complex analyses of sequence structure relationships, including: structure alignment of proteins, investigation of specific interaction motifs, studies of protein-protein and protein-DNA interactions, and protein super-families.
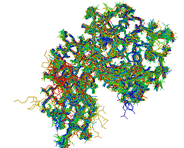
ModView is a program to visualize and analyze multiple bimolecular structures and/or sequence alignments. As a Netscape plug-in , it can be embed into Web pages and controlled by JavaScript objects on the page. It has wide range of tools to manipulate and analyze sequences and structures by interactive control.
Protein Structure Alignment method, TOPOFIT
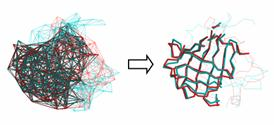
In the TOPOFIT method, similarity of protein structures is analyzed using three-dimensional Delaunay triangulation patterns derived from backbone representation. It has been found that structurally related proteins have a common spatial invariant part, a set of tetrahedrons, mathematically described as a common spatial sub-graph volume of the three-dimensional contact graph derived from Delaunay tessellation (DT).
We collaborate with Prof. SI Abarzhi on further development of the method.
Protein Structure Alignments Database, TOPOFIT-DB
TOPOFIT-DB (T-DB) is a protein structure alignment database of structural relations between all proteins in PDB.
(updated weekly, after PDB update; usually all new PDBs are aligned by Sat). Currently T-DB has some specific aims: 1). First to help researchers locate and analyze the structure neighbors found by the TOPOFIT method; including functional amino acids conservation, structural core analysis, and flexible region analysis. 2). Secondly, to give researchers, such as crystallographers, a portal through which one-to-all comparison of a newly determined structure against the entire PDB can be carried out. 3). Finally, through its online visualization software (Friend), provide users with the ability to quickly analyze the structural alignments stored in T-DB ( TEST ). All facets of the database are public and open to suggestions, comments and critiques.
Structural Exon Database, SEDB
SEDB is an application that allows users to retrieve the exon/intron organization of genes and map the location of the exon boundaries and the intron phase onto a multiple structure alignment. SEDB is linked with Friend, an integrated analytical multiple sequence/structure viewer, which allows simultaneous visualization of exon boundaries on structure and sequence alignments.
Structure SNP web server, StSNP
StSNP compare structural nsSNP distributions in many proteins or protein complexes. StSNP enables to researchers to map nsSNPs onto protein structures by comparative modeling of structure with nsSNPs by MODELLER and visualize their structural locations by using the multiple structure-sequence viewer Friend. Pathway information is provided from KEGG database. StSNP includes human nsSNPs.
We are interested in understanding cellular processes at molecular lever in atomic details to gain insight into functionality of molecular machines and answer what cause their malfunction, i.e. genetic and infection diseases. To address that we develop mathematical, statistical models, using physical and chemical laws, implement them in computer algorithms and methods, programs, databases toward solving problems in biology and medicine, specifically biomedical/biotech problems involving proteins, small molecules and DNA/RNA and their interactions. | |
| |
ilyinlab bioinformatics resources | Friend is a bioinformatics application designed for simultaneous analysis and visualization of multiple structures and sequences of proteins and/or DNA/RNA. The application provides basic functionalities such as: structure visualization with different rendering and coloring, sequence alignment, and simple phylogeny analysis, along with a number of extended features to perform more complex analyses of sequence structure relationships, including: structure alignment of proteins, investigation of specific interaction motifs, studies of protein-protein and protein-DNA interactions, and protein super-families. |
| ModView is a program to visualize and analyze multiple bimolecular structures and/or sequence alignments. As a Netscape plug-in , it can be embed into Web pages and controlled by JavaScript objects on the page. It has wide range of tools to manipulate and analyze sequences and structures by interactive control.
|
| Protein Structure Alignment method, TOPOFITIn the TOPOFIT method, similarity of protein structures is analyzed using three-dimensional Delaunay triangulation patterns derived from backbone representation. It has been found that structurally related proteins have a common spatial invariant part, a set of tetrahedrons, mathematically described as a common spatial sub-graph volume of the three-dimensional contact graph derived from Delaunay tessellation (DT). We collaborate with Prof. SI Abarzhi on further development of the method. |
| Protein Structure Alignments Database, TOPOFIT-DB TOPOFIT-DB (T-DB) is a protein structure alignment database of structural relations between all proteins in PDB. (updated weekly, after PDB update; usually all new PDBs are aligned by Sat). Currently T-DB has some specific aims: 1). First to help researchers locate and analyze the structure neighbors found by the TOPOFIT method; including functional amino acids conservation, structural core analysis, and flexible region analysis. 2). Secondly, to give researchers, such as crystallographers, a portal through which one-to-all comparison of a newly determined structure against the entire PDB can be carried out. 3). Finally, through its online visualization software (Friend), provide users with the ability to quickly analyze the structural alignments stored in T-DB ( TEST ). All facets of the database are public and open to suggestions, comments and critiques. |
| Structural Exon Database, SEDBSEDB is an application that allows users to retrieve the exon/intron organization of genes and map the location of the exon boundaries and the intron phase onto a multiple structure alignment. SEDB is linked with Friend, an integrated analytical multiple sequence/structure viewer, which allows simultaneous visualization of exon boundaries on structure and sequence alignments. |
| Structure SNP web server, StSNP StSNP compare structural nsSNP distributions in many proteins or protein complexes. StSNP enables to researchers to map nsSNPs onto protein structures by comparative modeling of structure with nsSNPs by MODELLER and visualize their structural locations by using the multiple structure-sequence viewer Friend. Pathway information is provided from KEGG database. StSNP includes human nsSNPs. |


Creating places that enhance the human experience.






